Das STZ EURO bietet Ihnen eine herstellerunabhängige Beratung und konzeptionelle Unterstützung möglichst bereits in der ersten Planungsphase, um für Sie die wirtschaftlichste Lösung für Ihre Reinraumtechnik zu erarbeiten.
Der Abscheidegrad (A) beschreibt, wie viel Prozent der aufgegebenen Partikel im Filter zurückgehalten werden. Er bildet das Gegenstück zum Durchlassgrad (D) und wird somit berechnet als A = 100% – D.
Akzeptanztest
Der Akzeptanztest wird durchgeführt, um festzustellen, ob ein System die definierten Kriterien (Akzeptanzkriterien) erfüllt. So soll der Kunde in die Lage versetzt werden, das System zu akzeptieren oder abzulehnen. Solche Tests können bereits sinnvoll sein, bevor eine Maschine oder Anlage ausgeliefert wird, siehe auch FAT und SAT. Die Akzeptanztests fungieren somit als Überprüfung der erforderlichen Eigenschaften und ob das System unter realen Einsatzbedingungen ordnungsgemäß funktioniert.
Aseptisch
Bedeutet “keimfrei” bzw. “ohne die Beteiligung von Erregern”.
Barrieresystem
Befindet sich zwischen dem Arbeiter und dem Produktionsbereich eine physische Abgrenzung, spricht man von einem Barrieresystem. Da Menschen zu den größten Risiken für eine Kontamination in einem Reinraum gehören, kann es sinnvoll sein, diese Bereiche voneinander zu trennen. Dies tut man beispielsweise mit Hilfe sogenannter Isolatoren oder RABS.
Betriebszustand
Ein Umgebungsmerkmal reiner Bereiche für die Herstellung steriler Produkte. Im Reinraum werden drei Betriebszustände unterschieden: „as built“, „at rest“ und „in operation“.
1. „as built“ = Bereitstellung; die Reinraumanlage (Decke, Wände, Boden, Lüftung usw.) ist vollständig installiert und funktionsfähig, aber es ist weder eine Inneneinrichtung vorhanden, noch ist Personal im Reinraum anwesend.
2. „at rest“ = Leerlauf; die Reinraumanlage ist voll funktionsfähig, die Inneneinrichtung ist vorhanden und das Equipment ist installiert. Das Personal ist jedoch noch abwesend und die Anlagen sind nicht in Betrieb bzw. in einem definierten Betriebszustand, der ohne Reinraumpersonal möglich ist.
3. „in operation“ = Fertigung; die Reinraumanlage ist voll funktionsfähig, die Inneneinrichtung ist vorhanden, das Equipment ist installiert und das Personal ist in der vorgesehenen Personenzahl anwesend. Die Anlagen im Reinraum fertigen Produkte in der vorgesehenen Betriebsart.
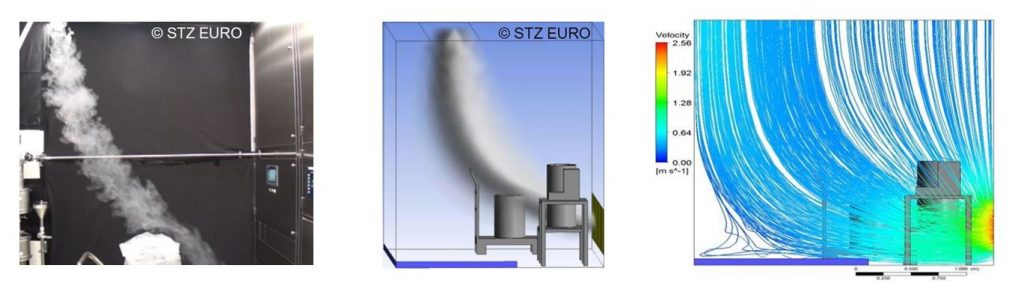
CFD-Simulation
„CFD“ steht für Computational Fluid Dynamics, zu Deutsch „Numerische Strömungsmechanik“. Sie ist vor allem bei der Planung, aber auch der Optimierung von Reinräumen, Lüftungs- und prozesstechnischen Anlagen wichtig. Mittels einer Computersimulation werden nicht nur die Luftströme, sondern auch Partikelkonzentrationen, Temperatur und weitere Größen berechnet und können gegebenenfalls optimiert werden.
Containment
Der englische Begriff „Containment“ ist nach VDI 2083-19 als Dichtheitsebene bzw. gasabgrenzende Ebene zu verstehen und ist somit die Hüllfläche um einen oder mehrere dichte Räume mit festgelegten Anforderungen. Anmerkung 1: Das Containment muss vor der Prüfung festgelegt und vereinbart werden. Anmerkung 2: Das Containment ist eine Fläche im Sinne einer Bilanzgrenze. Anmerkung 3: Im Sinne dieser Richtlinie können die Anforderungen an das Containment sowohl für eindringende wie auch ausströmende Kontaminanten definiert werden. Ein Containment kann z. B. ein Reinraum sein in dem mit hochaktiven Substanzen gearbeitet wird, die nicht in die Umgebung gelangen dürfen oder auch ein Isolator in den keine Verunreinigungen von außen eindringen dürfen.
Containmenttechnologie
Betrifft alle Maßnahmen (technisch, organisatorisch usw.), die erforderlich sind um das Schutzziel des Containments sicherzustellen.
DEHS
DEHS steht für Di-Ethyl-Hexyl-Sebacat, ein Fluid zur Erzeugung von Aerosolen für Filterintegritätstests und Erholzeitmessung.
Dichtheitsprüfung
Seit August 2018 existiert die Richtlinie VDI 2083-19, die einen Standard zu Planung, Durchführung und Klassifizierung von Dichtheitsprüfungen von Reinräumen festlegt. Die Dichtheit eines Containments wird seither durch eine Luftdichtheitsklasse (0-7) festgelegt, die mittels einer standardisierten Dichtheitsprüfung ermittelt wird.
Durchlassgrad
Der Durchlassgrad (D) ist das Verhältnis der abströmseitigen Partikelkonzentration (Reinluft CR) zur anströmseitigen Partikelkonzentration (Reinluft CROH) am Schwebstofffilter, ausgedrückt in %. Er beschreibt wie viel der aufgegebenen Partikel den Filter passieren. D = CR/CROH * 100%
Dynamisch
Dynamische Bedingungen beziehen sich auf eine Reinraumklassifizierung oder sonstige Prüfungen, wie beispielsweise Strömungsvisualisierung unter normalen Produktionsbedingungen (auch “in operation”genannt).
Eingriff (Intervention)
Ein Eingriff ist jede (aseptische) Manipulation oder Aktivität im kritischen Bereich, die vom Reinraumpersonal oder auch einem Roboter ausgeführt wird und dabei die Strömungsverhältnisse negativ beeinflussen kann.
Erholzeitmessung
Die Erholzeitmessung ist ein Prüfverfahren für Belüftungsanlagen im Reinraum. Durch die Erholzeitmessung kann die Aufreinigungsleistung des gesamten Raumes und die Effizienz der turbulenten Lüftung bewertet werden.
Zuerst wird der Raum mit einer definierten Aerosolkonzentration beaufschlagt.
Anschließend wird geprüft, ob die Anlage fähig ist, innerhalb einer begrenzten Zeitspanne den Partikelgehalt wieder auf das festgelegte Maß, entsprechend der Reinraumklasse, zu regulieren.
Bei korrekter messtechnischer Durchführung muss an jeder Stelle im Raum die gleiche Erholzeit gemessen werden. Die Effizienz der Lüftung kann als Wirkungsgrad angegeben werden. Dieser ist durch das Verhältnis von theoretischer zu gemessener Erholzeit zu bestimmen.
Wird per VDI 2083-3 festgelegt als Luft, die denselben Reinheitsgrad hat wie die aus den Schwebstofffiltern austretende Luft.
FAT
Steht für Factory Acceptance Test und bezeichnet allgemein die Werksabnahme. Im Gegensatz zum Site Acceptance Test (SAT); hier werden relevante Anlagen beim Hersteller vor Auslieferung aufgebaut und getestet.
FFU
Die Filter Fan Unit oder auch Filter-Ventilator-Einheit (FVE) ist ein elementarer Bestandteil der meisten Reinräume. In diesem Modul wird Luft aus dem Reinraum oder einem Plenum angesaugt, bei Bedarf gekühlt und danach durch einen Schwebstofffilter zurück in den Raum geblasen. Dadurch kann man dem reinen Bereich auf energieeffiziente Weise einen quasi partikelfreien und großen Luftvolumenstrom von ca. 1.600 m3/h pro Quadratmeter FFU-Fläche zuführen. Der reine Bereich kann sowohl eine TAV-Strömung, z. B. RABS, als auch ein Reinraum mit TVS sein.
Gleichgerichtete Strömung (Unidirectional Flow)
ist ein Luftstrom, der sich in robuster und gleichmäßiger Weise und mit ausreichender Geschwindigkeit in eine Richtung bewegt, damit zuverlässig alle Partikel aus dem kritischen Produktions- oder Testbereich verdrängt werden.
GMP
Bedeutet Good Manufacturing Practice, zu Deutsch „Gute Herstellungspraxis“. Sie umfasst Richtlinien und Regeln zur Herstellung von Arzneimitteln und Wirkstoffen, sowie gewissen Kosmetika und Lebensmitteln. Mit diesen Regularien soll sichergestellt werden, dass empfindliche Produkte eine konsistent hohe Qualität im gesamten Herstellungs- und Transportprozess aufweisen. Dies wird im Rahmen von Behörden,- Kunden und Selbst-Inspektionen regelmäßig überprüft. Da viele Arzneimittel in Reinräumen bearbeitet werden, müssen die in den GMP-Regularien beschriebenen Anforderungen an Reinraum- und Lüftungstechnik bereits bei der Planung befolgt werden. Ein besonders wichtiges Dokument für die sterile Herstellung von Arzneimitteln ist in diesem Zusammenhang der Annex 1 zum GMP-Leitfaden. Ergänzend dazu sind die anerkannten Regeln der Technik, wie ISO 14644 und VDI 2083 zu beachten.
H202-Dekontamination
Muss ein Containment oder Reinraum aus irgendwelchen Gründen mikrobiologisch dekontaminiert werden, ist die Behandlung mit Wasserstoffperoxid (H2O2) ein bewährtes Mittel. Hierbei wird das H2O2 verdampft und in einem Luftstrom dem Raum zugeführt oder im Raum als Aerosol freigesetzt. Der Raum wird damit gleichmäßig begast bzw. beaufschlagt. Bei entsprechender Konzentration wirkt das H2O2 auf den Oberflächen als starkes Oxidationsmittel. Auf diese Weise können Bakterien, Pilze, Viren und Sporen rückstandslos abgetötet und der Raum dekontaminiert werden. Dabei kommt es zu folgenden technischen Herausforderungen, die durch eine geeignete Planung und Ausführung aber problemlos beherrscht werden:
– Materialien/Oberflächen können beschädigt werden
– Es kann durch Leckagen in Wänden/Decken/Lüftungssystemen zu unzulässig hohen H2O2-Konzentrationen in der Umgebung des dekontaminierten Raums kommen und die gewünschte H2O2-Konzentration kann nicht erreicht werden
– Das Dekontaminations-Verfahren muss validiert und regelmäßig überwacht werden, damit es zuverlässig funktioniert
– Es sind erhöhte Anforderungen an die Lüftungs- und Automatisierungstechnik zu berücksichtigen
Isolator
Bezeichnet eine reine Zone mit starrer oder flexibler Einhausung. In der Regel sind Isolatoren nicht begehbar, der Zugriff erfolgt nur über Handschuhe. Ein Isolator besitzt eine eigene Luftversorgung mit Raumdruckregelung sowie ein integriertes Dekontaminationssystem mit Wasserstoffperoxid. Die reine Luft strömt meist über Doppelglasfenster zur integrierten Luftversorgung zurück.
Keimfreiheit/Asepsis
Unter Keimfreiheit oder Asepsis versteht man einen Kontrollzustand, bei dem in einem aseptischen Arbeitsbereich aseptische Arbeiten so durchgeführt werden, dass eine mikrobiologische Kontamination des ungeschützten Produkts ausgeschlossen wird.
Kontamination
Kontamination bezeichnet die Verunreinigungen chemischer, physikalischer oder mikrobiologischer Natur von Arbeitsbereichen, Einrichtungen, Maschinen, Werkzeugen, Materialien, Arbeitskleidung, der Haut, oder der Atemluft. Den Prozess der Beseitigung einer Kontamination nennt man Dekontamination.
Kritischer Bereich
Darunter wird üblicherweise der Bereich verstanden, an den die höchsten Reinheitsanforderungen gestellt werden, weil dort das Produkt oder produktberührende Oberflächen der Umgebungsluft ausgesetzt sind. Beispiel aseptische Vialsabfüllung: Offene Vials, Füllnadeln, Stopfen usw. Der kritische Bereich ist zu definieren.
Kritische Oberflächen (Critical Surfaces)
Dies sind Oberflächen, die mit sterilen Produkten, deren Behältern oder Verschlüssen in Kontakt kommen oder sie direkt beeinflussen können. Kritische Oberflächen werden vor Beginn des Herstellungsvorgangs sterilisiert, was während des gesamten Verarbeitungsverfahrens erhalten bleibt.
LF-Bereich
LF steht für „Laminar Flow“ und ist eine reine Zone innerhalb eines Reinraums mit TAV-Strömung. Bei LF-Bereichen strömt die Luft über Schwebstofffilter (Erstluft) in die reine Zone und wird durch Einhausungen (z.B. Lamellenvorhänge) zum kritischen Bereich geführt. Die Luft strömt im einfachsten Fall ohne messbaren Differenzdruck in den Reinraum über. Hier spricht man von einem konventionellen LF. LF-Bereiche können aber auch als Isolator, RABS, cRABS Sicherheitswerkbank o.Ä. ausgeführt werden.
Monitoring
Das Monitoring ist ein Überbegriff für die Überwachung von Vorgängen oder Prozessen. Für die Produktion im Reinraum bedeutet dies die ständige Erfassung physikalischer und prozesskritischer Größen, wie z. B. Temperatur, Feuchte, Raumdifferenzdruck, Luftgeschwindigkeit, Partikelkonzentration usw. Diese Werte werden ständig ausgewertet und protokolliert. Grenzwertüber- oder Unterschreitungen werden zeitnah erfasst, sodass in den Prozess rechtzeitig eingegriffen werden kann. Um Grenzwertverletzungen zu vermeiden, die häufig dazu führen, dass Produkte nicht freigegeben werden, ist es sinnvoll, engere Warngrenzen zu definieren, die ein noch früheres Eingreifen ermöglichen. So können negative Folgen vermieden werden.
MPPS
Steht für Most Penetrating Particle Size und bezeichnet die Partikelgröße im Abscheidegradminimum beim Nennvolumenstrom, das heißt die Partikelgröße, bei der die Fraktionsabscheidegradkurve ihr Minimum bzw. die Fraktionsdurchlassgradkurve ihr Maximum hat.
Nutzungsänderung
Es kommt vor, dass sich die Anforderungen an ein Containment oder Reinraum ändern, z.B. durch optimierte Herstellungsprozesse. Im Gegensatz zu herkömmlichen Produktionsbereichen können in einem Reinraum nicht einfach Gegenstände verschoben, entfernt oder hinzugefügt werden. Muss ein Reinraum in irgendeiner Form angepasst, modernisiert oder verändert werden, spricht man von einer Nutzungsänderung. Jede Nutzungsänderung muss weitsichtig geplant und durchgeführt werden. Hierbei sollten stets Experten hinzugezogen werden, um die wirtschaftlichste und sicherste Lösung zu finden.
Penetration
Die Penetration (P) ist gleichbedeutend mit dem Durchlassgrad (D) und wird ebenfalls in Prozent angegeben. P = D
Pneumatik
Das Wort Pneumatik bezeichnet den Einsatz von Druckluft in Wissenschaft und Technik zur Verrichtung mechanischer Arbeit.
Qualifizierung, prospektive
Mittels der Qualifizierung eines Reinraums oder einer Anlage wird dokumentiert nachgewiesen, dass Planung, Bau und Inbetriebnahme nach den entsprechenden normativen und benutzerspezifischen Vorgaben erfolgt sind. Bei der prospektiven Qualifizierung wird diese Aktion während der Planung (Designqualifizierung/DQ) begonnen und wird üblicherweise mit der Leistungs- oder auch Performancequalifizierung/PQ abgeschlossen. Die prospektive Qualifizierung oder Erstqualifizierung kommt üblicherweise bei neu zu beschaffenden Reinräumen und Anlagen zur Anwendung, d.h. bevor diese für die GMP-Produktion genutzt werden. Siehe auch retrospektive Qualifizierung.
Qualifizierung, retrospektive
Eine retrospektive Qualifizierung wird bei bereits in Betrieb genommenen Reinräumen und Anlagen durchgeführt, wenn diese bisher noch nicht qualifiziert waren aber bereits für die GMP-Produktion genutzt wurden. Die retrospektive Qualifizierung wird von den Behörden nicht mehr als geeignet betrachtet. Siehe auch prospektive Qualifizierung.
RABS
Restricted Access Barrier System. Der wesentliche Unterschied zu einem konventionellen LF-Bereich besteht in der Verwendung von Handschuhen für Routineeingriffe. Die reine Luft strömt zum umgebenden Reinraum über.
Reiner Bereich (Clean Area)
Ist festgelegt als ein Bereich mit definierten partikulären und mikrobiologischen Reinheitsstandards.
Reinraumklassen
Reinräume haben grundsätzlich die Aufgabe, eine Umgebung mit möglichst wenig Partikeln in der Luft zu ermöglichen. Sobald ein Reinraum konstruiert, gebaut, in Betrieb genommen und gereinigt worden ist, werden in diesem initial und regelmäßig wiederkehrend Partikelmessungen durchgeführt. Aus der Anzahl der in der Luft gemessenen Partikel ergibt sich die Reinraumklasse. Heutzutage gibt es hauptsächlich zwei Skalen für Reinraumklassen. Die erste betrifft die Einstufung allgemeiner Reinräume und wird festgelegt durch DIN EN ISO 14644-1. Diese teilt Reinräume in neun verschiedene Klassen (ISO 1- ISO 9) ein, wobei ISO 1 die höchste Klasse darstellt. In einem Reinraum der Kategorie ISO 1 dürfen sich in einem Kubikmeter Luft nur noch maximal 10 Partikel befinden, die größer oder gleich als 0,1 µm sind. In einem Reinraum ISO 9 dagegen dürfen sich noch 293.000 Partikel befinden, die eine Größe von 5,0 µm oder mehr aufweisen. Zwischen diesen beiden Extremen lässt sich jeder Reinraum messen und in eine der Klassen einteilen. Die andere Skala für Reinräume betrifft speziell die Herstellung steriler Arzneimittel und wird festgelegt durch den EU-GMP Leitfaden bzw. den zugehörigen Anhang 1 (Annex 1). Hier wird eine Aufteilung in vier Kategorien vorgenommen: A, B, C und D. Grund für diese Unterscheidung ist, dass bei der Herstellung pharmazeutischer Produkte neben den Partikeln auch die mikrobiologische Belastung der Luft und der Oberflächen relevant ist und daher in der Klasseneinteilung Berücksichtigung findet. Zudem muss in diesen Reinräumen sowohl der Ruhezustand als auch der Betriebszustand gemessen werden. Die Grenzwerte für Partikel sind hier dann auch nicht gleich, sondern liegen im operationellen Zustand, d.h. mit Anwesenheit von Personen und laufender Produktion für die Klassen B bis D deutlich höher. Grob übertragen lässt sich sagen, dass Reinräume (im Ruhezustand) der Klassen A und B hier etwa der ISO 5 Kategorie der oben beschriebenen Skala entsprechen. Ein Reinraum C entspricht ISO 7 und D entspricht ISO 8. Diese Werte einschließlich der Werte für den operationellen Zustand gehen aus nachfolgender Tabelle hervor. Übersicht Reinraumklassen A, B, C, D:
Zusammenhang zwischen GMP und ISO-Reinheitsklassen Auf Basis des aktuellen Annex 1 (2008)
Reinheitsklasse
Maximal erlaubte Zahl von Partikeln pro Kubikmeter
Ruhezustand
Betriebszustand
≥ 0,5µm
≥ 5µm
Entspr. ISO
≥ 0,5µm
≥ 5µm
Entspr. ISO
A
3520
20
5/4,8
3520
20
5/4,8
B
3520
29
5
352000
2900
7
C
352000
2900
7
3520000
29000
8
D
3520000
29000
8
Nicht festgelegt
Zusammenhang zwischen GMP und ISO-Reinheitsklassen Geplante Änderungen auf Basis des Entwurfs Annex 1 (02/2020) für die Klassifizierungsmessung
Reinheitsklasse
Maximal erlaubte Zahl von Partikeln pro Kubikmeter
Ruhezustand
Betriebszustand
≥ 0,5µm
≥ 5µm
Entspr. ISO
≥ 0,5µm
≥ 5µm
Entspr. ISO
A
3520
-n.a.
5
3520
-n.a.
5
B
3520
-n.a.
5
352000
-2900
7
C
352000
-2900
7
3520000
-29000
8
D
3520000
-29000
8
n.d.¹
-n.d.
–
Requalifizierung
Bei der Requalifizierung wird sichergestellt, dass sich die Anlage weiterhin in einem qualifizierten Zustand befindet. Es handelt sich dabei um eine periodische Überwachung bzw. Evaluierung. Die Zeitabstände und Kriterien für die Evaluation müssen begründet und festgelegt werden.
Schwebstofffilter
Bezeichnet Filter zur Abscheidung von Schwebstoffen aus der Luft. Sie werden nach aufsteigender Abscheidewirksamkeit in die Gruppen E, H und U eingeteilt:
Schwebstofffilter E (EPA – Efficiency Particulate Air Filter), das in eine der Klassen E10 bis E12 nach DIN EN 1822-1 eingestuft ist.
Schwebstofffilter H (HEPA – High Efficiency Particulate Air Filter), das in eine der Klassen H13 bis H14 nach DIN EN 1822-1 eingestuft ist.
Schwebstofffilter U (ULPA – Ultra Low Penetration Air Filter), das in eine der Klassen U15 bis U17 nach DIN EN 1822-1 eingestuft ist.
Simulation
Bei der Simulation wird versucht, ein möglichst realitätsnahes Nachbilden der Wirklichkeit zu erreichen. Die Simulation wird über spezielle Software digital durchgeführt. Durch Abstraktion soll so ein Modell geschaffen werden, an dem zielgerichtet experimentiert wird. Die daraus ableitbaren Ergebnisse werden dann auf das reale Problem übertragen. Nachbilden eines Systems mit seinen dynamischen Prozessen in einem experimentierfähigen Modell, um Erkenntnisse zu erlangen, die auf die Wirklichkeit übertragbar sind” ( VDI 3633 Blatt 1).
Simulationen gehören heute bereits in vielen Anwendungsgebieten zu einer bewährten Methode, um Produkte zu entwickeln, Konsequenzen besser voraussehen zu können und realitätsnahe Prognosen abzugeben. In der Reinraumtechnik wird die Simulation vor allem für das Ableiten von Luftströmungen und zur Vorbereitung von Inbetriebnahmen und (Re-)Qualifzierungsmaßnahmen eingesetzt.
Sorptionstrocknung
Bei dieser Trocknung kommen Adsorptionstrockner oder Trockenschränke zum Einsatz, die mit entsprechenden Trockenmitteln Feuchtigkeit entziehen. Der Vorgang funktioniert nach dem Sorptionsprinzip.
Steril
Als steril werden Gegenstände und Materialien bezeichnet, die möglichst frei von jeglichen lebenden Organismen, wie Bakterien, Pilzen und Viren sind. Eine absolute Keimfreiheit gibt es nicht, allenfalls eine Reduzierung der vorhandenen Mikroorganismen, die zu einer relativen Keimfreiheit führt. In der Praxis lässt sich die absolute Beseitigung von Mikroorganismen nicht nachweisen.
Strömungsvisualisierung
Um in einem Reinraum optimale Bedingungen für Produkte und Personen zu gewährleisten, müssen unter allen relevanten Betriebsbedingungen die Strömungsverhältnisse innerhalb des Raumes stimmen. Zu deren Überprüfung wird in Reinräumen eine Strömungsvisualisierung durchgeführt. Dies ist ein Testverfahren, bei dem geeigneter Prüfnebel in den reinen Bereich geleitet wird, dessen Ausbreitung dann visualisiert, videotechnisch aufgezeichnet und bewertet wird. Bei komplexen Anlagen wird nach einem Drehbuch vorgegangen. Das Testverfahren ist erstmalig umfangreich in der VDI 2083 Blatt 3 (Entwurf 2020) beschrieben. Vorbereitend zur Strömungsvisualisierung kann bereits in der Planungsphase eine Überprüfung und Optimierung der Strömung per CFD erfolgen.
TAV
Reine Zone innerhalb eines Reinraums, die Abkürzung steht für turbulenzarme Verdrängungsströmung.
TVS
Bezeichnet einen Reinraum mit Turbulenter Verdünnungsströmung.
Validierung
Die Validierung (lat. Valere – Wert sein, Wert haben) ist Erbringung eines dokumentierten Nachweises, der mit einem
hohen Grad an Sicherheit gewährleistet, dass ein Prozess stetig ein Produkt erzeugt, dass seine vorab festgelegten Spezifikationen und Qualitätskriterien erfüllt.
VDI 2083 Blatt 19
VDI 2083-19 ist eine Richtlinie, in der erstmals Dichtheitsanforderungen von Containments im Reinraumbereich klar und eindeutig beschrieben werden. Dabei werden nicht nur die Anforderungen definiert, sondern auch Prüf- und Messverfahren sowie Vorgehensweisen im Projektmanagement.
Anhand der neuen Richtlinie können für alle relevanten Containments, die Dichtheitsanforderungen definiert und messtechnisch überprüft werden.
Die Dichtheit des Containments wird zukünftig durch die Festlegung einer Luftdichtheitsklasse (0-7) und eines Bezugsdifferenzdrucks definiert.
Es steht mit VDI 2083 Blatt 19 eine ganzheitlich ausgerichtete und praxisorientierte Richtlinie zur Verfügung, die standardgemäße Vorgehendweise bei allen relevanten Projektphasen aufschlüsselt – von der Planung, dem Bau, der Qualifizierung bis hin zur Requalifizierung.